汉德-许勒尔-克思斯琴病


汉德-许勒尔-克思斯琴病
 流行病学
流行病学
病因
发病机制
临床表现

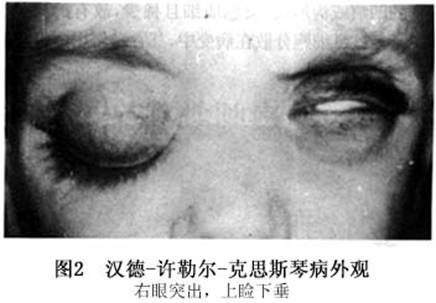 并发症
并发症
实验室检查
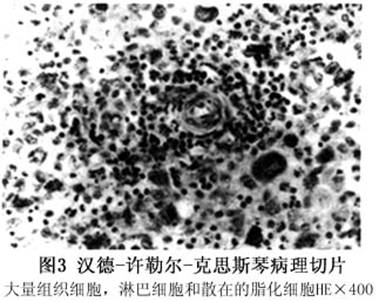 其他辅助检查
其他辅助检查
鉴别诊断
治疗
预后
预防

轻触这里
关闭目录